Can Rapamycin Prevent Alzheimer’s? What the Research Shows

For decades, the “amyloid hypothesis” has dominated neurobiology, yet drugs targeting plaque clearance have yielded mixed clinical success [1] [2]. A paradigm shift is now underway, focusing on geroscience: the idea that targeting biological aging itself is the most effective way to prevent Alzheimer’s disease (AD) [3]. At the center of this movement is rapamycin (sirolimus), an FDA-approved mTOR inhibitor that has consistently extended lifespan in every animal model tested [3] [4]. Emerging research suggests rapamycin’s true power may not be in treating advanced dementia, but in a precisely timed prevention strategy for high-risk groups, particularly APOE4 carriers [5] [6].
Rapamycin and Alzheimer’s disease
What rapamycin is and how it works
Rapamycin is a macrolide antibiotic discovered on Easter Island that functions as a potent, specific inhibitor of the mechanistic target of rapamycin (mTOR) [7] [8]. Within cells, rapamycin binds to the FKBP12 protein to allosterically inhibit mTOR complex 1 (mTORC1), which regulates nutrient sensing, cellular growth, and the essential cleanup process known as autophagy [3] [8] [9].
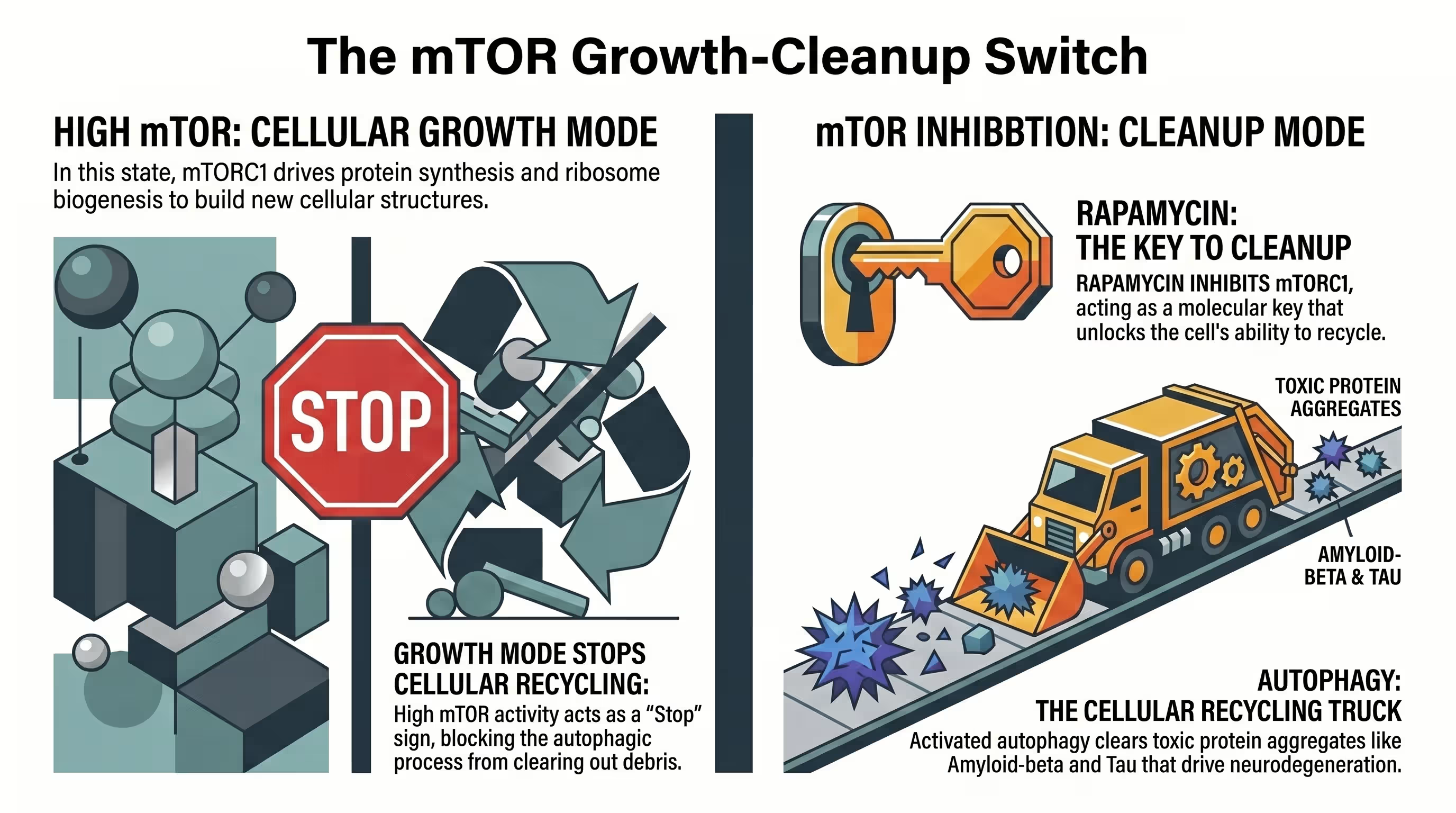
The discovery of rapamycin originated from the soil of Rapa Nui, where it was first identified as an antifungal agent before its immunosuppressive and anti-aging properties were uncovered [4] [7] [10]. In the human body, mTOR acts as a master nutrient sensor, integrating signals from insulin, growth factors, and amino acids to dictate whether a cell should grow or recycle damaged components [5] [8] [11]. mTORC1 inhibition by rapamycin shifts the cell from a “growth mode” to a “maintenance mode,” stimulating the lysosomal degradation of misfolded proteins [5] [11] [12].
Chronic over-activation of mTOR is a hallmark of the aging brain and is particularly elevated in the hippocampi of patients with Alzheimer’s disease [11] [12] [13]. By dampening this hyperactive signaling, rapamycin restores cellular homeostasis and facilitates the clearance of toxic aggregates like amyloid-beta and tau [8] [12] [14]. Beyond autophagy, rapamycin also targets other Hallmarks of Aging, such as mitochondrial dysfunction, cellular senescence, and chronic inflammation [3] [9].
Why mTOR inhibition matters for brain aging
mTOR inhibition matters because hyperactive mTOR signaling drives the accumulation of proteotoxic waste, suppresses mitochondrial efficiency, and reduces cerebral blood flow, all of which accelerate cognitive decline [12] [15]. By reducing mTOR activity to more youthful levels, rapamycin can restore synaptic plasticity, enhance neurovascular coupling, and maintain the integrity of the blood-brain barrier [5] [14] [15].
As the brain ages, the delicate balance between protein synthesis and protein degradation (proteostasis) begins to fail [12] [16]. This failure is largely mediated by mTORC1, which, when over-active, shuts down the “cleanup crew” (autophagy) while simultaneously promoting the synthesis of potentially toxic proteins [7] [17]. This create a “perfect storm” for neurodegeneration, where amyloid-beta (Aβ) plaques and neurofibrillary tau tangles suffocate neurons [5] [7].
mTOR inhibition has been shown to be one of the most effective and reproducible pharmacological approaches for increasing healthspan and delaying age-related pathologies in laboratory animals [3] [15]. In the aging brain, rapamycin-induced mTOR attenuation prevents and reverses cognitive and cerebrovascular dysfunction in multiple AD models [11] [18]. This neuroprotection is multifaceted: it restores cerebral glucose uptake, maintains white matter structural integrity, and activates endothelial nitric oxide synthase (eNOS) to “open up” blood vessels and improve brain oxygenation [8] [13] [19].
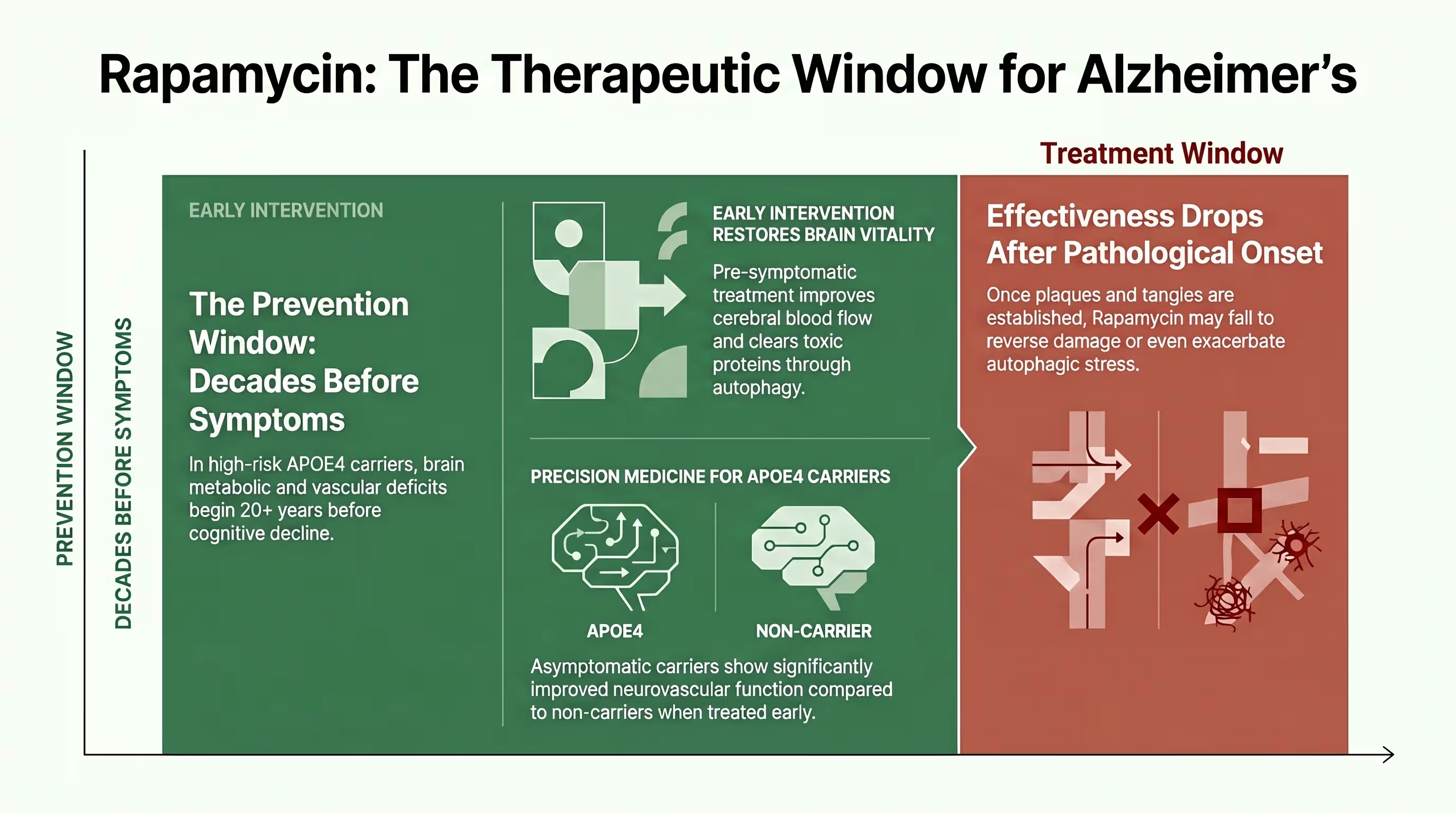
Why Alzheimer’s prevention is the key question
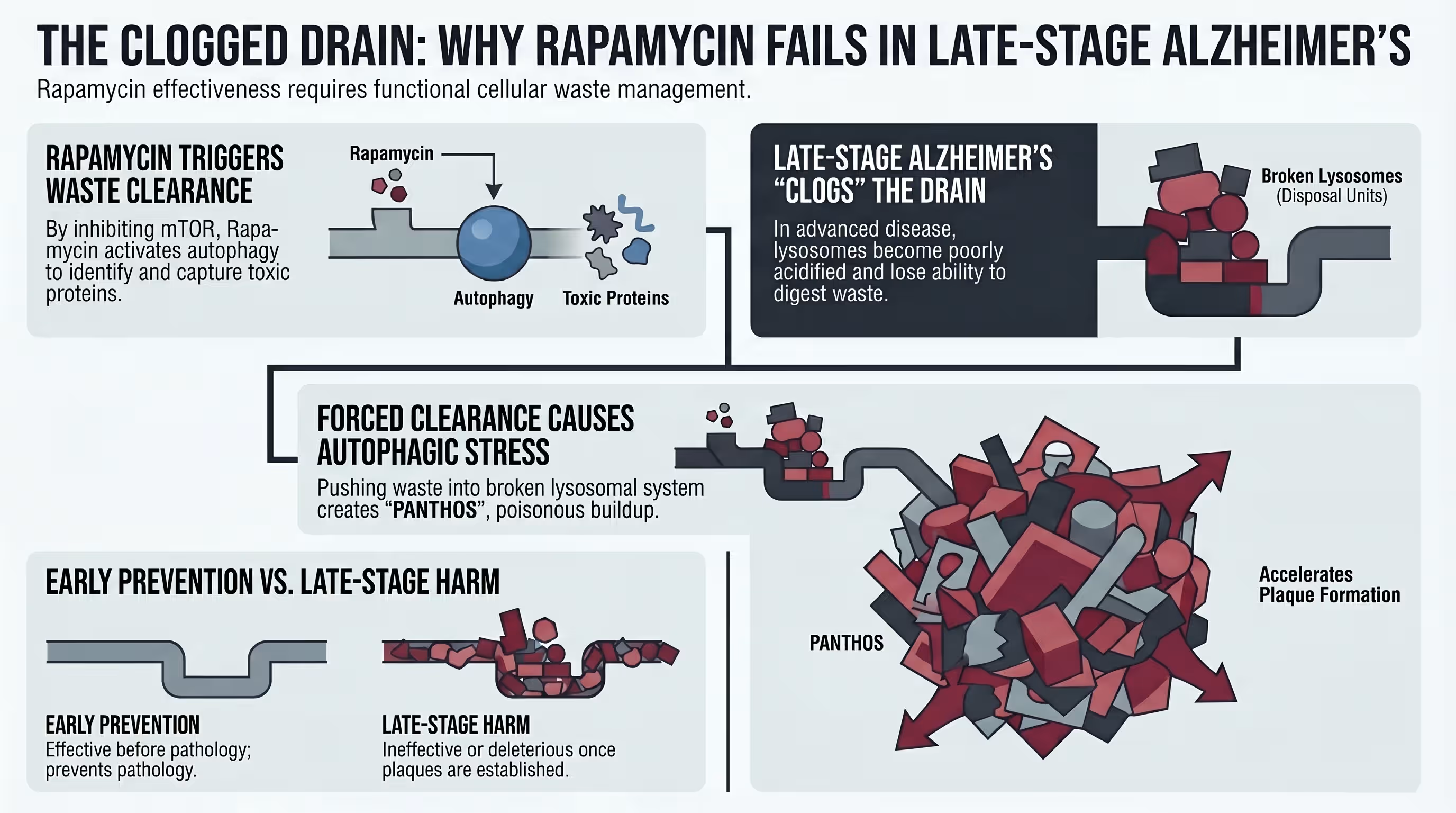
Alzheimer’s prevention is the key question because established neuropathology, such as mature plaques and tangles, appears largely resistant to mTOR-induced clearance in late stages [7] [8] [16]. Research indicates a specific “therapeutic window” where rapamycin is highly effective when given prophylactically but can become ineffectual—or even harmful—once the brain’s lysosomal system is too damaged to handle induced autophagic stress [7] [8] [16].
The standard of care for Alzheimer’s has historically focused on “rescuing” patients who already exhibit symptoms, yet by the time dementia is diagnosed, amyloid plaques have often been developing for two decades [1] [16]. Preclinical data from 3xTg-AD mice shows that lifelong rapamycin treatment initiated before pathology results in an 86% reduction in cortical tangles and preserved memory [4] [7]. However, when the same treatment was started at 15 months of age (after plaques were established), it had no effect on pathology or cognitive deficits [7] [16].
This stage-dependency highlights the critical importance of early intervention [4] [8] [20]. If the brain’s “drain” (the lysosomal system) is already clogged with undigested material, simply turning on the “faucet” of autophagy via rapamycin can lead to autophagic stress [8] [16]. Therefore, the most promising application of rapamycin lies in its use as a biological shield for asymptomatic individuals at high genetic risk, such as APOE4 carriers, who exhibit vascular and metabolic deficits decades before symptoms arise [5] [13] [15].
What the research shows
Preclinical findings in mouse and cell models
Preclinical research across diverse AD models demonstrates that rapamycin significantly reduces Aβ loads, inhibits tau hyperphosphorylation, and prevents spatial memory loss [2] [3]. Studies have found that rapamycin-treated mice perform in the same ballpark as healthy “wild-type” animals on complex cognitive tasks like the Morris Water Maze [4] [17] [19].
The breadth and depth of preclinical data for rapamycin in AD are perhaps greater than for any other potential therapy currently being investigated [3]. In specific models of tauopathy, such as P301S mice, rapamycin not only reduced forebrain tau pathology but also significantly diminished astrogliosis (brain inflammation/scarring) [21] [22]. In models expressing human Aβ and APOE4 (E4FAD mice), 16 weeks of rapamycin feeding normalized body weight and restored hippocampal cerebral blood flow (CBF) [13] [15].
Behavioral assessments have confirmed these physiological improvements [8] [13] [23]. Rapamycin-treated AD mice show a significantly higher “recognition index” in novel object recognition tests, independent of their genotype [13]. Furthermore, systemic rapamycin treatment has been shown to protect the perforant pathway—a critical brain circuit involved in memory—from tau-mediated neurodegeneration and synapse loss [4] [22]. These findings suggest that rapamycin targets multiple facets of AD pathophysiology simultaneously [5] [22].
Evidence for reduced amyloid and tau pathology
Rapamycin reduces pathology by upregulating the autophagic clearance of misfolded proteins and restoring the integrity of the blood-brain barrier (BBB) to facilitate the export of toxic waste [5] [14] [19]. In long-term mouse studies, early intervention with rapamycin reduced Thioflavin-S positive plaques by over 50% and lowered insoluble tau levels in the forebrain [7] [21].
The mechanism for pathology reduction is primarily tied to mTORC1 inhibition, which induces the formation of autophagosomes that capture and degrade protein aggregates [17]. Research has shown that rapamycin significantly decreases the accumulation of soluble Aβ42 and phosphorylated tau (p-tau) epitopes [4] [7] [11]. By enhancing the activity of the P-glycoprotein (P-gp) transporter at the BBB, rapamycin helps “pump” Aβ out of the brain and into the peripheral circulation for disposal [5] [13].
Crucially, rapamycin’s effect on tau is not limited to mere clearance; it also modulates the phosphorylation pathways that lead to tangle formation in the first place [21]. In a study of P301S tau transgenic mice, both long-term and short-term rapamycin treatment significantly lowered tau hyperphosphorylated at critical sites like AT8 and AT100 [21]. However, researchers emphasize that while rapamycin alleviates pathology, it does not necessarily reduce the total generation of tau, reinforcing its role as a clearance-enhancer rather than a direct protein synthesis inhibitor in this context [21] [22].
Evidence for improved brain metabolism and synaptic function
Rapamycin improves brain metabolism by enhancing mitochondrial oxidative metabolism, increasing neuronal TCA cycle rates, and preserving levels of excitatory neurotransmitters like glutamate [15]. Using advanced neuroimaging, researchers found that rapamycin-treated APOE4 mice restored cerebral glucose uptake (CMRGlc) and synaptic density to levels seen in healthy wild-type controls [2] [19] [23].
Brain metabolic deficits, specifically glucose hypometabolism, are now recognized as playing a more critical role in the progression of cognitive impairment than Aβ and tau levels alone [5] [15]. In young, asymptomatic APOE4 carriers, rapamycin treatment was found to restore neuronal mitochondrial function and increase total glutamate-glutamine neurotransmitter cycling (Vcycle) [5] [15]. This metabolic preservation is essential because glucose utilization provides the energy required for neurotransmission and memory formation [19] [23].
Furthermore, rapamycin has been shown to protect the synapse—the point of communication between brain cells [22]. In AD models, mTOR hyperactivation often leads to “leaky” synapses and impaired synaptic plasticity [10] [12]. Rapamycin mitigates this by improving the communication between neurons and preserving the density of synaptic connections in the hippocampus [2] [4] [22]. By bridging metabolic restoration with neurovascular protection, rapamycin ensures that neurons have both the fuel and the structural integrity to function optimally [5] [19].

Human trial evidence
Prevention trial in cognitively normal APOE4 carriers
A groundbreaking human pilot study demonstrated that just 4 weeks of low-dose rapamycin (1 mg/day) significantly increased cerebral blood flow (>15%) in cognitively normal middle-aged APOE4 carriers [4] [6] [24]. These carriers also showed an enrichment of beneficial gut microbiome populations (e.g., Butyricicoccus) and a reduction in systemic inflammatory cytokines [6].
The first-in-human trial evaluating rapamycin for AD prevention focused on individuals aged 45–65 who carry the APOE4 allele but remain cognitively normal [6]. Using 3T MRI-based arterial spin labeling, researchers found that rapamycin specifically reversed the early vascular deficits associated with the ε4 allele [4] [6] [24]. The increase in CBF was most pronounced in female carriers, reaching up to 45% in critical cognitive regions like the inferior temporal gyrus [6] [24].
Metabolic shifts were also observed: treated carriers saw an increase in Glycerol-3-phosphate (G3P), signaling a move toward healthy lipid metabolism and enhanced neuronal energy support. This trial highlighted a genotype-specific response, as non-carriers showed stable CBF and different inflammatory patterns compared to carriers. Importantly, the treatment was safe and well-tolerated, with participants even reporting subjective improvements in mood and energy levels [6].
Therapeutic trial in mild cognitive impairment and early Alzheimer’s
The CARPE_DIEM and REACH trials, which enrolled older adults already diagnosed with MCI or early AD, found that 1 mg/day of rapamycin for 8-12 weeks was safe but yielded mixed results [10] [11]. In these late-stage symptomatic patients, rapamycin was not detected in the cerebrospinal fluid (CSF), and researchers observed a concerning numerical increase in AD biomarkers like p-tau 181 and GFAP [10] [11] [20].
In contrast to the prevention data, the open-label Phase 1 trial in symptomatic patients (n=10, mean age 74) did not observe meaningful cognitive improvements over the short treatment duration. The lack of detection of rapamycin in the CSF—despite its high lipophilicity—suggests that at a 1 mg/day dose, the drug may not reach therapeutic concentrations in the diseased human brain at this advanced stage [10] [11] [14].
The increase in CSF glial fibrillary acidic protein (GFAP) and neurofilament light (NfL) potentially indicates increased autophagic stress or a reactive biological response in a brain where neurodegeneration is already active[10]. Furthermore, participants saw an increase in hemoglobin A1c and systolic blood pressure, markers that require close monitoring during mTOR inhibition [11] . These results underscore that late-stage intervention presents significantly higher hurdles and different safety considerations than early prevention [8] [11] [20].
Stage-dependent results and why timing matters
Research reveals a striking stage-dependency: rapamycin reduces inflammation and increases blood flow in young/middle-aged pre-symptomatic groups but increases inflammatory and AD biomarkers in older, symptomatic patients [8] [11]. This suggests that rapamycin may have markedly divergent biological effects depending on whether it is used for prevention or treatment [20].
The “timing gap” is perhaps the most critical takeaway from current rapamycin research [20]. Preclinical models consistently show that rapamycin works best when started before the formation of plaques and tangles. If treatment is initiated too late, rapamycin may actually exacerbate pathology by triggering more cellular waste than the aging lysosomes can clear [7] [8] [16].
This creates a paradox: the individuals most desperate for treatment (those with active dementia) may be the least likely to benefit, while those who are currently healthy (young APOE4 carriers) represent the ideal target for intervention [5] [15]. Consequently, clinical trial designers are now pivoting toward middle-aged populations, using neuroimaging to identify the precise window when brain metabolic and vascular deficits first appear—often 20 years before a clinical diagnosis [13] [14] [23].
APOE4 and risk
Why APOE4 carriers are the highest-risk group
The APOE4 allele is the strongest genetic risk factor for late-onset AD, increasing risk by 3-12 times compared to non-carriers [5] [19]. APOE4 carriers develop neurovascular and metabolic impairments—such as reduced cerebral blood flow and impaired blood-brain barrier integrity—decades before memory loss or amyloid deposition manifest [5] [13].
Apolipoprotein E is a lipid transporter that exists in three variants: ε2 (protective), ε3 (neutral), and ε4 (high risk) [5]. Even in their 40s and 50s, cognitively normal APOE4 carriers exhibit regional hypometabolism in brain areas vulnerable to AD [5]. This is partly because APOE4 proteins have a structural preference for binding to large, triglyceride-rich lipoproteins, which can form neurotoxic complexes and impair lipid turnover [13].
APOE4 also drives early vascular uncoupling and oxidative stress [8] [13]. These carriers exhibit accelerated pericyte degeneration, leading to a “leaky” blood-brain barrier that allows peripheral toxins to infiltrate the brain [23]. Because these physiological changes precede irreversible cognitive decline, they represent the primary therapeutic target for prevention strategies [13] [23].
What genotype-dependent effects suggest
Research indicates that rapamycin effects are highly genotype-dependent: it restores CBF and enhances Aβ clearance in APOE4 carriers but primarily impacts glycolytic pathways and reduces anxiety in APOE3 non-carriers [5] [13] [15]. This suggests that rapamycin activates different metabolic signaling pathways (e.g., SREBP1 vs HIF1α) based on the individual’s genetic background [13] [15].
In studies comparing E4FAD and E3FAD mice, rapamycin normalized free fatty acid levels and restored neurotransmitter balance specifically in the APOE4 genotype [5] [13]. In contrast, APOE3 mice—who did not have baseline vascular deficits—responded to rapamycin by increasing glycolysis and inhibitory neurotransmission, similar to the effects seen with caloric restriction [13] [15].
These differential results emphasize that we cannot assume a “one-size-fits-all” response to mTOR inhibition [5] [13] [15]. While rapamycin acts as a “rescue agent” for the vascular and metabolic damage caused by APOE4, it acts more as a “metabolic optimizer” in healthy non-carriers [13] [15]. This highlights the necessity of using genetic profiling to tailor dosing and predict outcomes in future human trials [5] [6] [15].
| Genotype | Cerebral Blood Flow Change | Gut Microbiome Change | Treatment Duration | Physiological Measure |
| APOE4 Carrier | Significantly increased ( $>15\%$ , primarily in females); restored hippocampal CBF and normalized to levels similar to E3FAD | Enriched with Bacteroides salyersiae , Bacteroides stercoris , and Butyricicoccus sp. | 4–16 weeks | Restored BBB function (P-gp activity), reduced $A\beta$ retention, reduced plasma cytokines, preserved neurotransmitters, and improved mitochondrial function |
| APOE4 Non-Carrier | No significant change (stable) | Enriched with Intestinimonas sp. and unclassified Ruminococcaceae genus | 4–16 weeks | Enhanced spatial recognition memory, reduced anxiety, reduced brain glycolysis, and promoted inhibitory neurotransmission |
Why precision medicine matters
Precision medicine matters because identifying the timeframe of brain physiological changes allows for intervention with the right dose at the right time [13] [15] [23]. Using non-invasive MRI and PET imaging to track CBF and glucose metabolism enables clinicians to assess an individual’s personal response to rapamycin and adjust therapy accordingly [2] [13] [14] [15].
The emerging field of geroscience-informed intervention recognizes that biological aging is not a uniform process [3] [5] [11]. Precision medicine for AD prevention requires integrating genetic testing (APOE status), biomarker discovery (p-tau, GFAP, NfL), and therapeutic innovation [6] [11] . For example, female APOE4 carriers may require different dosing regimens than male non-carriers due to sex-hormone-driven differences in mTOR regulation [6] [13].
By using multimodal neuroimaging, researchers can detect vascular and metabolic changes that precede cognitive dysfunction by decades [13] [15] [23]. This allows for a proactive rather than reactive approach. Combining rapamycin with other targeted treatments—such as anti-inflammatory agents or tau modulators—could optimize efficacy while ensuring that treatment is personalized to the specific needs and risk profiles of each patient [5] [11] [23].
Biological mechanisms
Effects on cerebral blood flow
Rapamycin restores cerebral blood flow by acting as a nitric oxide-dependent vasodilator through the activation of endothelial nitric oxide synthase (eNOS) [5] [13] [19]. In human carriers, short-term rapamycin treatment resulted in regional CBF increases exceeding 20% across cognitive centers like the frontal and parietal lobes [15] [24].
Cerebrovascular dysfunction is one of the earliest detectable changes in Alzheimer’s patients. Inadequate perfusion leads to nutrient starvation, oxidative stress, and the reduced ability of the brain to “wash out” toxic metabolic byproducts [8] [14] [23]. Rapamycin preserves vascular density and restores “neurovascular coupling”—the process by which the brain adjusts local blood flow based on neuronal activity [3] [16] [19].
In preclinical models, this restoration was associated with reduced accumulation of Aβ and cerebral amyloid angiopathy [5] [19]. Human data confirms that these vascular benefits can be achieved with low, safe doses [4] [6]. By “opening up the pipes” of the brain’s vascular network, rapamycin ensures that neurons have the necessary oxygen and glucose to maintain memory and learning functions [5] [23].

Effects on inflammation and cytokines
Rapamycin functions as a “cenomorphic” agent, suppressing the senescence-associated secretory phenotype (SASP) and reducing peripheral inflammatory cytokines such as MCP-1 and Tle-2 [6] [9]. It dampens the NF-κB inflammatory switch and promotes immune tolerance through the enhancement of T-regulatory cell function [5] [6] [8].
Chronic inflammation is now recognized as a central mechanism of AD, not just a byproduct [7] [14]. Rapamycin-induced mTOR inhibition reduces the production of proinflammatory markers like IL-1β and IL-6, which otherwise facilitate amyloid and tau deposition [4] [5]. Interestingly, while high daily doses of rapamycin are immunosuppressive, low intermittent doses have been shown to actually improve immune function in older adults, such as enhancing vaccine responses [5] [6] [19].
Furthermore, rapamycin’s anti-inflammatory effects extend to the brain’s resident immune cells: microglia [13] [14]. While rapamycin generally attenuates microglial activation, researchers caution that long-term use could potentially diminish the ability of microglia to “uptake” and clear established plaques in very late stages. This reinforces the strategy of using rapamycin as a “preventive shield” to keep inflammation low rather than a late-stage fire extinguisher [8] [20].
Effects on metabolism and gut microbiome
Rapamycin shifts metabolism toward efficient lipid turnover, reduces free fatty acid levels, and enriches beneficial gut bacteria that produce anti-inflammatory short-chain fatty acids (SCFAs) like butyrate and propionate [6] [13]. It also lowers red cell distribution width (RDW), a systemic marker of biological aging and oxidative stress [6].
The gut-brain axis is a critical pathway for Alzheimer’s risk [25]. In human APOE4 carriers, 4 weeks of rapamycin enriched microbiome populations such as Bacteroides salyersiae and Butyricicoccus, which support the integrity of the gut and the blood-brain barrier. These SCFA shifts are highly associated with improved brain metabolism and reduced systemic neuroinflammation [6] [15].
Metabolically, rapamycin reduces “lipotoxicity” by modulating lipid synthesis pathways (like SREBP1/PPARγ) [15]. This normalization of lipids, including polyunsaturated fatty acids, explains why rapamycin can stabilize body weight and improve energy metabolism in high-risk groups [13]. By targeting the blood, the gut, and the vessels simultaneously, rapamycin offers a multifaceted defense against the metabolic collapse that defines Alzheimer’s progression [5] [6].
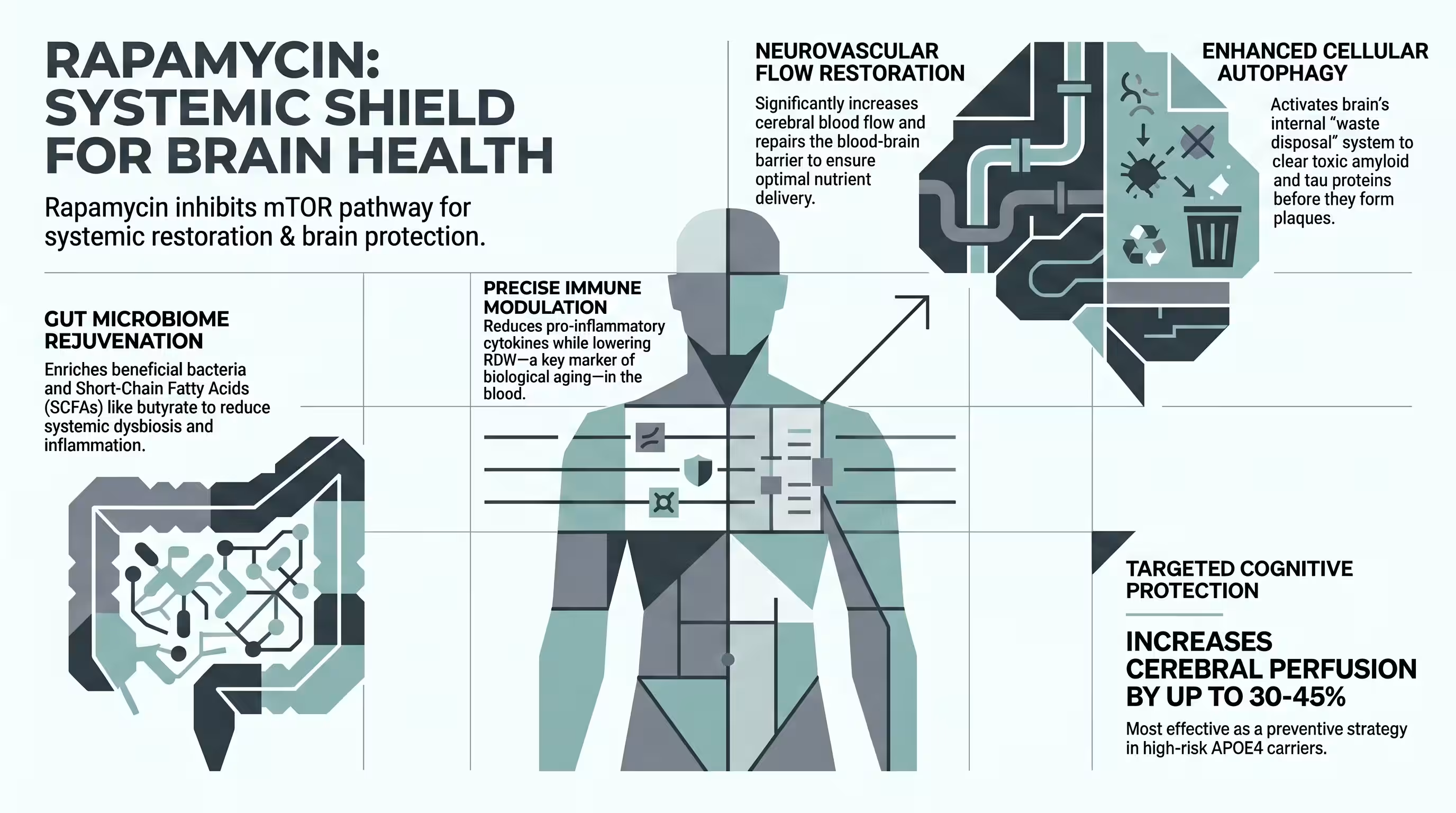
Effects on amyloid, tau, and neurodegeneration
Rapamycin mitigates neurodegeneration by abrogating tau-mediated neurotoxicity at its earliest stages and enhancing the autophagic degradation of Aβ [12]. Preclinical studies show a 92% reduction in tau-driven synapse loss and a significant reduction in the trans-synaptic expansion of human tau expression [22].
Neurodegeneration in AD is often initiated by the loss of the perforant pathway, a circuit that connects the entorhinal cortex to the hippocampus. Systemic rapamycin treatment has been shown to protect these neurons and their synapses from early-stage tauopathy [4] [22]. By preventing “synaptotoxicity”—where protein aggregates weaken the connections between brain cells—rapamycin maintains the brain’s cognitive “wiring” [8] [12].
While rapamycin is not a cure-all, its ability to target both the “hallmarks” (amyloid/tau) and the “drivers” (aging/metabolism) makes it a unique candidate for modifying the disease course [12] [14]. Laboratory results suggest that by stabilizing neurotransmitter levels and preventing neuronal apoptosis (cell death), rapamycin could transform Alzheimer’s from an inevitable outcome of aging into a pharmacologically manageable condition [8] [11].
| Feature | Prevention Trial (APOE4 carriers) | Therapeutic Trial (MCI/Early AD) |
|---|---|---|
| Cerebral Blood Flow | Significant increase (>15%) | Not assessed/Reported |
| AD Biomarkers | Stable (No change in p-tau/Aβ) | Increased (p-tau 181, NfL, GFAP) |
| Inflammation | Reduced cytokines (MCP-1) | Increased plasma cytokines |
| Metabolism | Enriched SCFAs (Butyrate) | Increased HbA1c |
| Primary Goal | Vascular/Metabolic restoration | Safety and CSF penetrance |
| Dose/Duration | 1mg daily for 4 weeks | 1mg daily for 8-12 weeks |
Table: Prevention vs. Therapeutic Human Trials
Conclusion: Is Rapamycin the Answer for Alzheimer’s?
Current research indicates that rapamycin is a potent preventive agent, not a “rescue” drug for advanced dementia.
Key Takeaways:
- Prevention is the Primary Goal: It works best when initiated before significant plaque and tangle formation.
- APOE4 Advantage: Carriers of the highest-risk gene see immediate improvements in brain blood flow and energy metabolism.
- Multimodal Defense: It protects the brain through autophagy, vascular restoration, and gut-brain modulation.
- Late-Stage Risks: Symptomatic patients may face “autophagic stress” and biomarker increases, requiring extreme caution.
- Precision Dosing: Weekly intermittent dosing appears to maximize safety while providing the neurovascular benefits seen in trials
If you are a middle-aged APOE4 carrier or interested in geroscience-based prevention, consult a physician specializing in longevity medicine. Never begin a rapamycin regimen without professional oversight, as personalized dosing and baseline monitoring (lipids, glucose) are essential for safety.

Current research suggests rapamycin is not a treatment for established AD and may actually be ineffectual or harmful in late stages.
Yes, rapamycin is highly lipophilic and has been detected in the brains of animal models. However, its penetrance in healthy human brains at low doses is still being debated.
There is no officially approved dose, but longevity studies typically use low weekly doses (e.g., 5-10 mg) to minimize side effects.
At high daily doses, yes. However, at low intermittent doses, it has been shown to actually improve immune response in the elderly.
Rapashop offers high quality medical grade rapamycin available at different tablet doses.
No. While both target aging pathways, metformin primarily targets AMPK/blood sugar, while rapamycin directly targets mTOR/autophagy.
Short-term trials show safety, but the long-term risk of metabolic shifts (like insulin resistance) requires ongoing physician monitoring.
Resource links
Rapamycin and Alzheimer's disease: Time for a clinical trial?
Rapamycin Responds to Alzheimer’s Disease: A Potential Translational Therapy
Matt Kaeberlein on Rapamycin Longevity Series | Lessons learned from 2 decades of Rapamycin research
Rapamycin treatment for Alzheimer’s disease and related dementias: a pilot phase 1 clinical trial
Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration
APOE genotype-dependent pharmacogenetic responses to rapamycin for preventing Alzheimer's disease
Rapamycin’s Anti-Alzheimer’s Promise: An Autophagy Story
Blazing a trail for the clinical use of rapamycin as a geroprotecTOR
mTOR: Alzheimer's disease prevention for APOE4 carriers
Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice